
AI+Human Translation in Pharma: Ensuring Compliance and Safety
- Feb 22
- 16 min read

Every Regulatory Affairs Manager knows the pressure of ensuring that every document submitted to the European Medicines Agency reflects both linguistic accuracy and regulatory compliance. With pharmaceutical documentation required in multiple languages across the European Union, even a single translation mistake can expose your company to audit findings or patient safety risks. By embracing a structured AI+Human hybrid translation model, you gain both the speed to meet compressed timelines and the auditability that satisfies regulators, providing a defensible, compliant process from source language to inspection-ready records.
Table of Contents
Key Takeaways
Point | Details |
AI+Human Hybrid Translation | This method ensures compliance through a structured workflow with SME review and terminology governance, suitable for regulated environments. |
Audit Trail | Each stage of the hybrid translation process generates an auditable record, crucial for regulatory inspections and compliance verification. |
Terminology Consistency | Utilizing Translation Memories and Term Bases enhances terminology control, mitigating risks associated with mistranslations. |
Speed and Efficiency | AI+Human hybrid translation offers faster turnaround times compared to traditional methods, improving operational efficiency across multilingual documentation. |
Defining AI+Human Translation in Pharma
AI+Human translation in pharma is not machine translation with post-editing. It is a structured, auditable hybrid workflow where artificial intelligence generates regulatory-grade translations under strict terminology governance, followed by certified subject-matter expert (SME) review for accuracy, compliance, and clinical safety.
This distinction matters operationally. In regulated environments like the European Union or FDA submissions, the difference between machine translation and AI+Human hybrid translation directly impacts your defense during an inspection.
Why the Definition Matters in Regulated Pharma
Pharmaceutical translations cross clinical, regulatory, and manufacturing domains simultaneously. A single submission dossier may include clinical trial protocols, safety data, manufacturing instructions, and pharmacovigilance reports—each with different terminology demands and risk profiles.
Machine translation (MT, neural MT, or public SaaS engines) processes text without context about pharmaceutical terminology standards or your organization’s approved term bases. The output is often literal and inconsistent.
AI+Human hybrid translation begins with asset integration. Your Translation Memory ™ and Term Bases (TB) load first, constraining the AI system to use approved terminology before the machine even generates output.
The research on collaborative translation models confirms this approach: AI excels at processing speed and breadth, while humans refine context and specialized knowledge, particularly in regulated sectors where terminology precision is non-negotiable.
Let’s map the core workflow.
The Four-Step AI+Human Hybrid Workflow
Asset integration — Load client Translation Memories ™ and Term Bases (TB) to enforce terminology consistency.
LLM generation — Proprietary AI system produces target language output constrained by client terminology and style guidance.
SME review — Certified subject-matter expert (medical professional, clinical translator, pharmacist) reviews for technical accuracy, regulatory compliance, and contextual nuance.
Quality assurance — QA aligned to ISO 17100 and ISO 18587 standards, with sector-specific checks (MDR, GCP, ICH guidelines).
This sequence is auditable. Each step produces a record: TM match reports, AI generation logs, SME sign-off, and QA metrics. Regulators can trace every decision.
Here is a summary of the critical workflow stages for AI+Human translation in pharma:
Stage | Main Activity | Outcome | Compliance Focus |
Asset Integration | Load TM & TB | Terminology consistency | Regulatory standards enforced |
LLM Generation | AI creates translation | Draft output matches client assets | Ensures terminology control |
SME Review | Expert checks translation | Technical and regulatory validation | Clinical accuracy guaranteed |
Quality Assurance | ISO-aligned QA checks | Auditable records generated | Inspection-ready process |
Why This Matters for EU Regulatory Affairs Managers
Under GDPR, your translation vendor’s data processing must be documented. Under MDR (Medical Device Regulation), clinical documentation must demonstrate control over terminology and accuracy. Under GxP (Good Manufacturing Practice), translation workflows must be validated and reproducible.
A hybrid model with SME oversight satisfies all three because:
Data sovereignty — If your provider hosts on EU infrastructure with private encryption, no data leaves your control.
Terminology governance — TM and TB assets remain yours; AI enforces them automatically.
Auditability — Every translation step is logged, traceable, and defensible during inspection.
Compare this to generic machine translation: no terminology control, no SME oversight, no audit trail. That approach exposes you to critical meaning errors in safety-critical text.
The regulatory difference is concrete: Hybrid AI+Human translation with SME review and terminology governance can be defended during an FDA inspection or EMA audit as a validated, controlled process. Public machine translation cannot.
How AD VERBUM Implements the Hybrid Model
AD VERBUM’s proprietary LangOps System combines all four steps into a single workflow. Client TM and TB load first, restricting the AI system to approved terminology before generation. A certified medical or clinical translator then reviews for accuracy and compliance, followed by ISO 17100 and ISO 18587 QA. The result is terminology-consistent, compliant, auditable output delivered 3x to 5x faster than traditional translation.
For EU-based regulatory affairs teams, the infrastructure detail matters: AD VERBUM operates on private EU servers, not public cloud platforms. That means no reliance on third-party SaaS providers, no data residency ambiguity, and GDPR compliance by design.
Key Operational Takeaways
AI+Human hybrid translation is a structured, auditable workflow with SME oversight and terminology governance.
Generic machine translation lacks both controls and is indefensible in regulated submissions.
The workflow produces an audit trail: TM matches, AI logs, SME sign-off, QA metrics.
For EU regulatory teams, data sovereignty and terminology control are non-negotiable operational requirements.
Hybrid translation can be validated and defended during inspections; machine translation cannot.
Pro tip: Before piloting any AI translation service, request documentation of their QA process, SME credential standards, and data processing agreements (especially GDPR compliance). If they cannot produce an audit trail linking terminology assets to final output, the service is not suitable for regulatory submissions.
Multilingual Document Demands Across Pharma Lifecycle
Pharmaceutical organizations operate globally. That simple fact creates a massive translation burden that extends across every phase of drug development, from laboratory discovery through post-market surveillance.
Your company does not translate for one market. You translate for dozens simultaneously, each with distinct regulatory requirements, terminology standards, and submission timelines. A clinical trial protocol written in English must reach investigators in Japan, Germany, Brazil, and Australia—each in the local language, each with identical meaning.
This is not a nice-to-have. It is a regulatory mandate.
The Documentation Footprint
Pharmaceutical product lifecycles require multilingual documentation spanning discovery, clinical trials, manufacturing, and post-authorization phases. Each phase generates distinct document types with specific translation demands.
Drug Discovery and Preclinical
Laboratory reports, chemistry data, safety assessments in source language.
Translation needed for: regulatory pre-submission meetings, investor communications, international research partnerships.
Clinical Trial Phase
Investigator’s Brochures (IB), clinical trial protocols, informed consent forms (ICF), safety reports.
Translated into every country where sites operate; ICF must match local regulatory language standards precisely.
Manufacturing and Compliance
Batch records, process validation, standard operating procedures (SOPs), quality control documentation.
Must be available in languages of all manufacturing facilities and regulatory jurisdictions.
Post-Market Surveillance
Pharmacovigilance reports, periodic safety update reports (PSUR), adverse event summaries.
Distributed to national regulators in their required languages; errors directly impact patient safety decisions.
Regulatory Submissions
Complete dossier translations for EMA, FDA, PMDA (Japan), NMPA (China), ANVISA (Brazil), and dozens of other authorities.
Each submission must conform to local formatting, terminology, and submission language requirements.
Why Translation Accuracy is a Patient Safety Issue
Pharmaceutical translation errors are not cosmetic. A mistranslation in a pharmacovigilance report might obscure a safety signal. An error in manufacturing instructions could compromise product quality. A mistranslated dosing section in clinical trial materials could harm participants.

When you submit to regulators, your translations carry the same legal weight as your source documents. The EMA and FDA treat translation quality as evidence of your company’s control over product information.
Consider the scale: A typical dossier submission spans 10,000 to 50,000 pages. If translated for five major markets, that is 50,000 to 250,000 pages of regulatory text that must be consistent, accurate, and auditable.
The burden is not just volume—it is velocity. Regulatory timelines compress across markets. You may have weeks to translate and validate submissions across 15 languages while maintaining terminology consistency and managing quality assurance.
Common Bottlenecks in Multilingual Pharma Workflows
Terminology inconsistency — Different translators or systems render the same medical term differently across languages, creating audit findings.
Timeline compression — Markets demand simultaneous submissions; sequential translation becomes impossible.
Regulatory variability — Each country’s health authority has distinct submission language requirements; one-size-fits-all translation fails.
Quality verification — Traditional review cycles cannot keep pace with volume; errors slip through.
Data sovereignty — Sensitive clinical and manufacturing data must stay within compliant infrastructure; many translation services cannot guarantee this.
Why AI+Human Hybrid Translation Addresses This Burden
The volume of multilingual pharma documentation is too large for traditional translation alone. Yet the safety and regulatory stakes are too high for fully automated machine translation.
AI+Human hybrid translation processes high volume at controlled quality by combining speed with expertise. The AI system generates baseline translations constrained by your terminology assets (Translation Memories and Term Bases), then certified subject-matter experts review for accuracy and compliance.
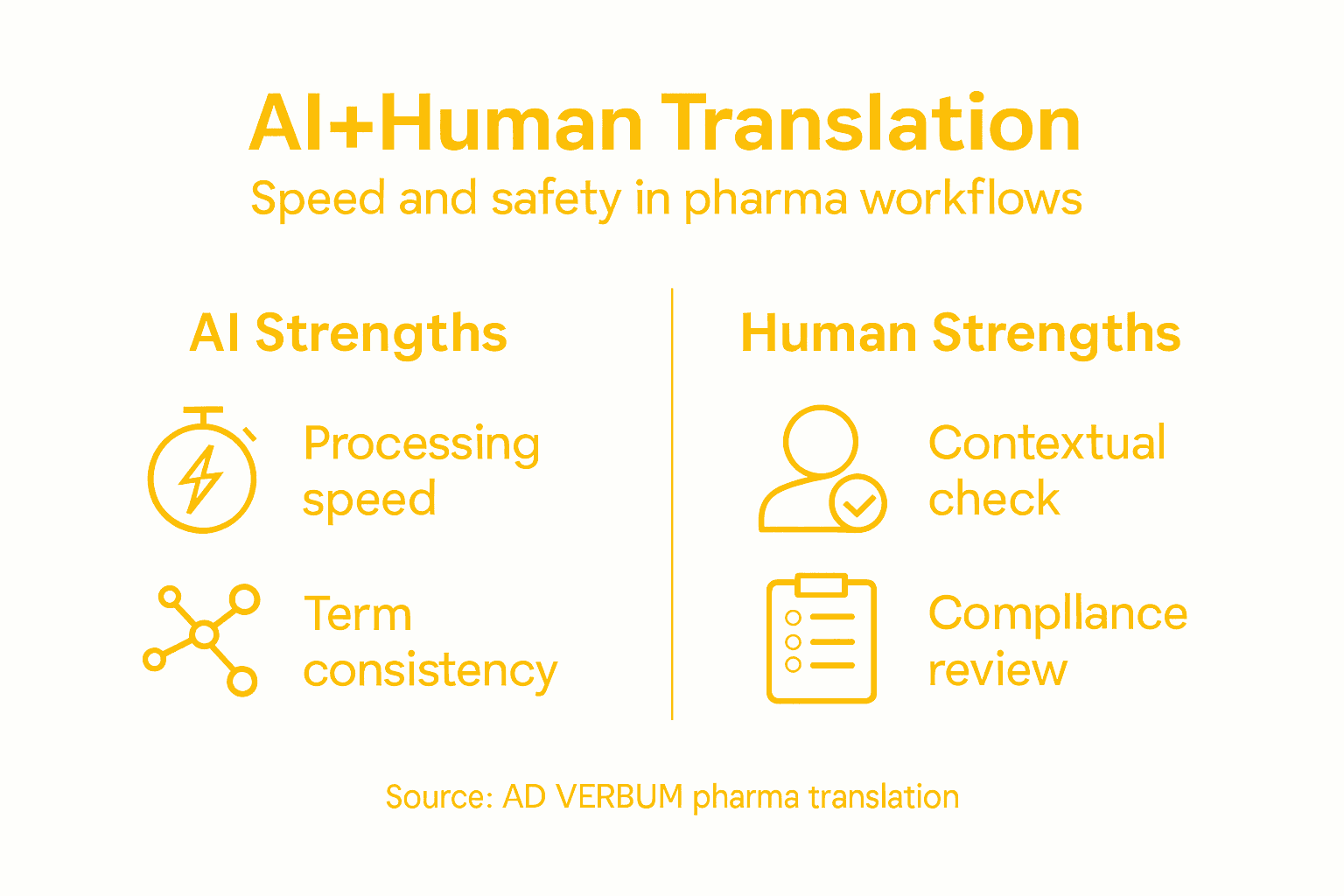
The result: faster turnaround than traditional translation, with audit trail and regulatory defensibility that machine translation cannot match.
Pro tip: Before entering a new market, audit your existing translation assets (TMs and Term Bases) for that language. If gaps exist, invest in populating them before you scale—clean terminology data is your foundation for consistent, compliant translations across all subsequent submissions.
Regulatory Risks: Translation Errors and Compliance
A single mistranslation in a regulatory submission can trigger inspection findings, delay approvals, or worse—compromise patient safety. Translation errors in pharmaceutical documentation are not clerical mistakes. They are compliance violations with legal and clinical consequences.
Regulators treat translation quality as direct evidence of your company’s control over product information. A poor translation suggests poor quality systems overall.
What Regulators Actually Look For
During FDA or EMA inspections, investigators examine your translation processes with specific focus areas:
Terminology consistency — Does “adverse event” translate the same way across all documents in a given language?
Technical accuracy — Do translated dosing instructions match the source exactly, without omission or addition?
Regulatory compliance — Does the translation conform to the target country’s submission language requirements?
Traceability — Can you produce an audit trail showing who translated, reviewed, and approved each document?
Version control — If a source document was updated, can you prove the translation was updated too?
Failure in any of these areas results in an inspection observation (Form 483 in FDA terminology). Repeated failures lead to warning letters and enforcement action.
Real Consequences of Translation Errors
Safety signal obscuration. A pharmacovigilance report mistranslates a symptom description. The national regulator in another country misses a safety signal and does not initiate a recall investigation. Patients are harmed.
Manufacturing control loss. A batch record is translated with incorrect chemical concentrations. The manufacturing team operates out of specification without knowing. The batch is released and distributed.
Clinical trial integrity. An informed consent form is mistranslated. Participants do not understand the true risks. The trial data becomes invalid; the regulatory submission is rejected.
Regulatory delay. A dossier submitted with inconsistent terminology across languages triggers requests for clarification. Approval timelines slip by months while you scramble to fix translations.
Compare major regulatory risks from translation errors in pharmaceutical submissions:
Risk Area | Potential Impact | Example Scenario |
Patient Safety | Harm from miscommunication | Wrong dosing in clinical trial |
Product Quality | Batch release errors | Incorrect chemical concentrations |
Regulatory Approval | Approval delays or rejection | Dossier with terminology inconsistencies |
Compliance Evidence | Inspection findings | Lack of audit trail for revisions |
Translation errors are compliance failures, not communication hiccups. Regulators view poor translation as evidence of inadequate quality systems and risk management.
Common Error Categories in Pharma Translation
Negation errors — “Do not” becomes “Do” due to translation system limitations; critical safety instructions flip meaning.
Abbreviation mishandling — Acronyms translated literally instead of adapting to target language conventions; “API” (Active Pharmaceutical Ingredient) mistranslated as a generic computing term.
Dosing unit mistakes — Milligrams converted to grams or vice versa; units of measurement translated incorrectly.
Context collapse — Technical terms translated without understanding pharmaceutical context; a term that has one meaning in general medicine becomes ambiguous in oncology.
Version drift — Source document updated but translated version not synchronized; regulatory submission contains obsolete information.
Why Traditional Translation Alone Is Insufficient
Manual translation workflows struggle to maintain consistency and speed at scale. A single translator working on a 20,000-page dossier will inevitably introduce inconsistencies. Multiple translators working in parallel risk different terminology choices for the same medical concept.
Yet you cannot afford to slow down. Regulatory windows close. Competitive pressure accelerates.
This is where ensuring compliant translations for regulated sectors becomes operationally critical. Translation quality cannot be an afterthought or a cost center—it must be a control point integrated into your quality management system.
AI+Human Hybrid as Risk Mitigation
The hybrid model addresses translation error risk by enforcing consistency at the machine level and expertise at the human level. Your Translation Memory and Term Bases load first, constraining the AI system to use only approved terminology.
A certified medical translator then reviews the output for accuracy, regulatory compliance, and contextual nuance. QA checks align to ISO 17100 and ISO 18587 standards, producing an auditable record.
The result: terminology consistency across all documents, faster turnaround than traditional translation, and a defensible audit trail.
Pro tip: Before your next regulatory submission, conduct a pre-submission terminology audit. Identify all key medical and technical terms, verify they are translated consistently across your source materials, and confirm your translation vendor understands the approved terminology in each target language—this single step prevents the majority of translation-related inspection findings.
AI+Human Hybrid vs. Other Translation Approaches
Not all translation methods are equal in regulated environments. Your choice between traditional translation, machine translation, and AI+Human hybrid directly impacts compliance risk, turnaround time, and audit defensibility.
Understanding the trade-offs is critical. Each approach has distinct strengths and failure modes in pharmaceutical contexts.
Traditional Human Translation
How it works: A qualified translator (ideally with pharmaceutical background) translates the document manually, then a reviewer checks for accuracy.
Strengths:
High contextual understanding and nuance.
SME knowledge applied throughout the process.
Audit-friendly when translators are qualified and documented.
Weaknesses:
Slow. A 10,000-page dossier takes months, not weeks.
Expensive. You pay for every hour of human labor.
Inconsistency risk. Multiple translators may render the same term differently without strict TM governance.
Bottleneck. Translator availability constrains your schedule.
Traditional translation works for small, low-urgency documents. It fails at scale and speed.
Machine Translation (MT) and Neural Machine Translation (NMT)
How it works: Software processes text algorithmically (MT) or using neural networks (NMT). Output is fast and cheap. No human review is built in.
Strengths:
Very fast. Translates thousands of pages in hours.
Very cheap. Minimal labor cost.
Handles volume easily.
Weaknesses:
No terminology governance. Inconsistent terminology across documents.
Context blindness. Cannot distinguish between homonyms or apply domain knowledge.
Negation errors. Critical safety instructions flip meaning (“do not administer” becomes “administer”).
No SME review. Errors pass uncaught.
Indefensible in regulated submissions. Regulators expect evidence of human expertise and control.
Public MT systems (Google Translate, DeepL for general use) are built for consumer speed, not pharmaceutical precision. Using them for regulatory submissions without post-editing is a compliance violation waiting to happen.
AI+Human Hybrid Translation
How it works: Translation Memory and Term Bases load first, constraining the AI system to approved terminology. An AI engine generates baseline output, then a certified subject-matter expert reviews for accuracy, compliance, and contextual nuance. QA checks validate against standards (ISO 17100, ISO 18587, MDR).
Strengths:
Speed. 3x to 5x faster than traditional translation.
Consistency. Hybrid approaches combining AI efficiency with human judgment produce superior outcomes in specialized pharmaceutical contexts where precision is critical.
Terminology control. TM and TB enforce approved terminology automatically.
Cost-effective. AI handles volume; humans focus on high-value review.
Auditable. Every step produces a record: TM matches, AI logs, SME sign-off, QA metrics.
Defensible. Regulators accept hybrid workflows as validated, controlled processes.
Weaknesses:
Requires initial TM and TB investment. Clean terminology data is essential.
Depends on SME availability. Your review timeline is constrained by qualified reviewer capacity.
Higher initial setup cost than pure MT.
Side-by-Side Comparison
Attribute | Traditional | Machine Translation | AI+Human Hybrid |
Speed | Slow (weeks/months) | Very fast (hours) | Fast (days) |
Consistency | Moderate (without TM) | Low | High (TM-enforced) |
Regulatory defensibility | High (if SME-qualified) | Very low | High |
Terminology governance | Manual | None | Automatic (TM/TB) |
Audit trail | Document-dependent | Minimal | Complete |
Cost | High | Very low | Moderate |
Scalability | Limited | Unlimited | High |
For EU regulatory affairs teams: Hybrid AI+Human translation is the only approach that simultaneously meets speed, consistency, compliance, and audit requirements across multilingual pharma documentation.
When to Use Each Approach
Traditional translation: Small documents (under 5,000 words), highly specialized terminology with no existing TM, one-time projects where speed is not critical.
Machine translation alone: Never acceptable for regulated pharma submissions. Acceptable only for internal communications or preliminary market research without regulatory impact.
AI+Human hybrid: Clinical trial protocols, regulatory dossiers, manufacturing documentation, pharmacovigilance reports—any document that must be accurate, consistent, and compliant at scale.
Pro tip: Before committing to any translation service for regulatory submissions, request their QA methodology and ask specifically how they handle negation, abbreviations, and dosing units. Request a sample translation of a short, complex paragraph from your field. Poor performance on these three categories is a red flag that the service lacks pharmaceutical expertise.
Piloting AI Translation: Practical Steps for Pharma
Starting an AI translation pilot does not mean moving immediately to regulatory dossiers. Strategic organizations begin with low-risk content, build confidence, and scale gradually. This staged approach minimizes disruption while building internal expertise and validating your vendor’s capabilities.
A successful pilot produces measurable outcomes: reduced turnaround time, consistent terminology, and an auditable process that regulators will accept. It also identifies gaps in your current infrastructure—particularly your Translation Memories and Term Bases.
Phase 1: Assess Scope and Select Content
Start small. Identify 2-3 documents that are:
Low regulatory risk — Internal SOPs, training materials, or non-critical manufacturing documentation rather than clinical trial protocols or regulatory submissions.
Terminology-heavy — Documents where consistency matters but the stakes are not submission-level yet.
Representative — Content that reflects the document types you will eventually translate at scale.
Manageable volume — 5,000 to 15,000 words per document to complete the pilot in 4-6 weeks.
Avoid choosing documents you have never translated before. You need baseline quality to measure improvement.
Phase 2: Prepare and Integrate Assets
This step is critical and often underestimated. Before translation begins, audit your existing Translation Memory ™ and Term Base (TB):
TM health check — Is your TM populated with approved pharmaceutical terminology? Does it reflect your company’s style preferences? Remove obsolete or incorrect entries.
TB completeness — Are key medical terms defined? Do they include context notes explaining when to use term A versus term B?
Gap identification — What terminology does your TM lack? Populate it before the pilot.
Clean assets are your foundation. Dirty TM data will degrade the pilot and waste SME review time.
Phase 3: Execute Pilot Translation
Practical AI translation pilots require rigorous human post-editing to ensure accuracy and compliance. The workflow is straightforward:
Upload source documents and TM/TB to your translation vendor’s system.
AI system generates baseline translations constrained by your terminology assets.
SME reviewer (medical professional or clinical translator) checks for accuracy, regulatory compliance, and contextual fit.
QA process validates against ISO 17100 and ISO 18587 standards.
Deliver and document the final translation with full audit trail.
Request that your vendor provide:
TM match reports (percentage of text matched to existing terminology).
AI generation logs (transparency into how the system processed your content).
SME sign-off (proof that a qualified person reviewed the output).
QA metrics (defects found and corrected).
Phase 4: Measure and Evaluate
After completion, measure concrete outcomes:
Turnaround time — How many days from submission to final delivery? Compare to your historical average for traditional translation.
Terminology consistency — Did the same medical term translate identically across all documents? Pull a list of key terms and verify.
Rework required — What percentage of SME feedback required actual corrections versus minor refinements?
Cost per word — Calculate total cost divided by word count (source + target languages).
Audit readiness — Can you produce a complete audit trail showing TM matches, AI output, SME review, and QA sign-off?
A successful pilot shows 3x to 5x faster turnaround, 95%+ terminology consistency, and <5% rework rate.
Phase 5: Escalate to Higher-Risk Content
Once you are confident, move to moderately regulated content: pharmacovigilance reports, manufacturing batch records, or clinical safety summaries. Use the same workflow but with enhanced QA.
Only after successful completion of moderate-risk content should you consider regulatory dossier submissions. That escalation requires additional validation and documentation.
Key Success Factors
Sponsor executive buy-in — Pilot success requires time investment from regulatory affairs and quality teams.
SME continuity — Use the same reviewer across the pilot to build consistency and learning.
Transparent vendor communication — Your translation service must provide complete audit trails and explain QA findings clearly.
Realistic timelines — Pilots take 8-12 weeks end-to-end, including planning, execution, and evaluation.
Piloting is not a shortcut—it is a discipline. Organizations that rush from pilot to regulatory submissions without proper escalation often encounter compliance gaps that auditors will flag.
Pro tip: During your pilot, document every question your SME reviewer raises and every correction they make. This feedback loop becomes invaluable for refining your TM and Term Base before you scale to full regulatory submissions—and it gives you a compelling narrative for regulators about how seriously you control translation quality.
Strengthen Your Pharma Translations with AD VERBUM’s AI+Human Hybrid Solution
Pharmaceutical translation demands absolute precision, strict terminology control, and full regulatory compliance. The risks of inconsistent or irresponsible machine translation are high, especially under GDPR, MDR, and FDA requirements. If you are looking to bridge speed with accuracy while maintaining an auditable, compliant process, AD VERBUM’s proprietary LangOps System offers a structured AI+Human hybrid workflow trusted by life sciences professionals.
Our approach integrates your Translation Memories and Term Bases first to enforce terminology consistency. Then a proprietary EU-hosted LLM generates context-sensitive translations that certified subject-matter experts review for technical accuracy, regulatory compliance, and clinical nuance. This process is backed by ISO 17100 and ISO 18587 aligned quality assurance — ensuring your documentation is inspection ready. With over 25 years of experience and a global network of 3,500+ linguists, AD VERBUM uniquely meets the strict demands of regulated pharmaceutical content across 150+ languages.
Take control of your translation risk and accelerate your multilingual submissions with a process designed for compliance and audit defense. Discover how AD VERBUM Specialized AI Translation can transform your pharma workflows.
Ready to secure compliant, terminology-governed translations supported by human expertise and AI efficiency Visit AD VERBUM Contact today to discuss your specific pharma requirements and start your journey toward safer, compliant multilingual documentation. Ensure your next regulatory submission is backed by translation quality you can trust.
Frequently Asked Questions
What is AI+Human translation in pharma?
AI+Human translation in pharma is a structured hybrid workflow that combines artificial intelligence-generated translations with certified subject-matter expert (SME) reviews to ensure accuracy, compliance, and clinical safety.
How does AI+Human translation differ from traditional machine translation?
Unlike traditional machine translation, AI+Human translation uses terminology governance and integrates client-approved Translation Memories and Term Bases to produce contextually accurate outputs verified by SMEs, making it suitable for regulatory submissions.
Why is terminology consistency important in pharmaceutical translations?
Terminology consistency is crucial because pharmaceutical documents require precise language and accurate translations across multiple, high-stakes submissions; inconsistent terminology can lead to regulatory issues and pose patient safety risks.
What are the main steps involved in the AI+Human hybrid translation workflow?
The key steps include asset integration (loading TM and TB), AI generation (creating translation outputs), SME review (validating accuracy), and quality assurance (ensuring compliance with ISO standards), resulting in an auditable process suitable for regulatory inspections.
Recommended
