
What Changes on May 28 When EUDAMED Becomes Mandatory
- May 16
- 9 min read

On May 28, 2026, EUDAMED transitions from a voluntary system to a legal obligation under MDR and IVDR. What changes on May 28 when EUDAMED becomes mandatory is not a documentation update. It is a shift to continuous digital reporting where non-compliance means immediate loss of market access, not a warning letter. Commission Decision (EU) 2025/2371 triggered a six-month transition period starting November 27, 2025, ending precisely on May 28. If your organization is not registered, your data is not in the system, and your translations are not certified, you are not selling in the EU.
Table of Contents
Key Takeaways
Point | Details |
May 28 is a hard deadline | Four EUDAMED modules become legally mandatory on May 28, 2026, with no grace period for non-compliant manufacturers. |
Six modules, two timelines | The first four modules are mandatory in May 2026; Vigilance and Clinical Investigations are expected to follow around 2027. |
Translation is a compliance control | Device labeling data, vigilance reports, and clinical summaries require certified translation before EUDAMED upload. |
Two LAAs are the minimum | Each organization must maintain at least two Local Actor Administrators to prevent total system access loss. |
Data remediation takes longer than expected | Internal ERP and product master data alignment can take 12 to 18 months, making early action non-negotiable. |
The six EUDAMED modules and what becomes mandatory
Understanding the EUDAMED implementation timeline requires separating the two rollout phases. The May 28 EUDAMED deadline covers four modules. The remaining two follow later.
Actors module
The Actors module is the entry point for everything else. Manufacturers, authorized representatives, importers, and other economic operators must register their organization and receive a Single Registration Number (SRN). Without an SRN, no device registration is possible. Each organization must designate Local Actor Administrators (LAAs) who manage user accounts and data access. At least two LAAs per organization are required because the last active LAA cannot be terminated, and losing access to the sole administrator account creates a total operational block.
UDI/Devices module
This module captures device registration data tied to Unique Device Identification. Manufacturers must submit the Basic UDI-DI, device identifiers, intended purpose, classifications, and technical dimensions. Many organizations discover at this stage that their internal ERP systems lack the structured fields EUDAMED requires, such as EMDN codes. Data cleaning and alignment can take 12 to 18 months before submissions are possible, which means organizations that have not started remediation are already behind.
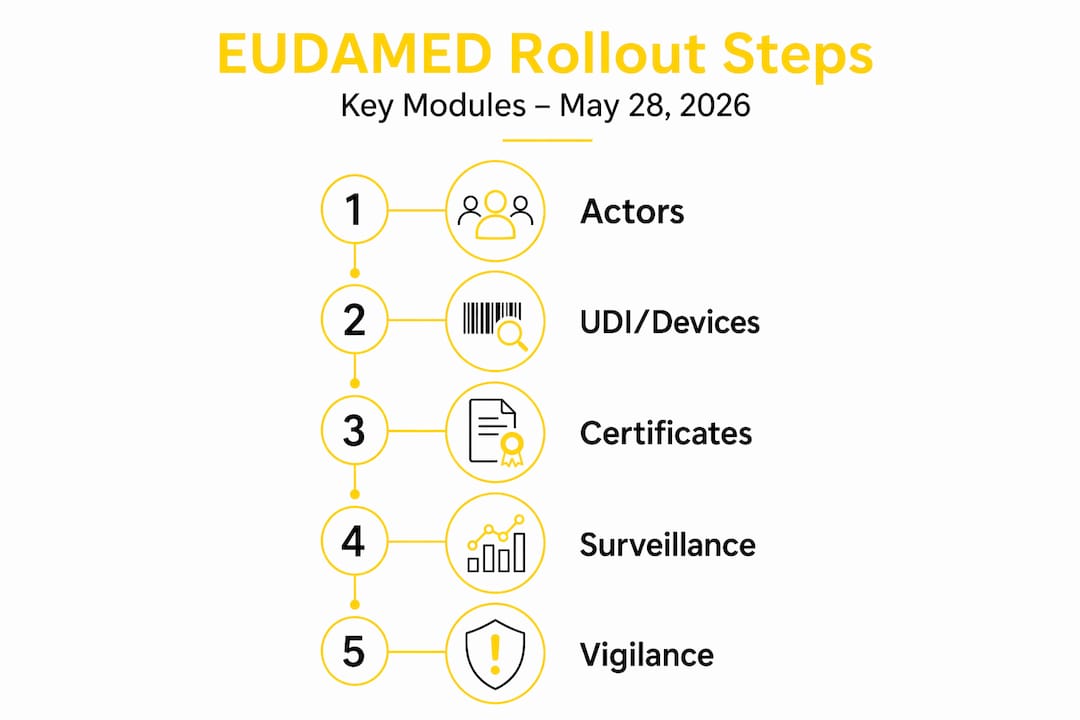
Notified Bodies and Certificates module
Notified Bodies upload conformity assessment certificates directly into EUDAMED. Manufacturers do not control this upload, but they are responsible for ensuring the data in their own submissions is consistent with what the Notified Body records. Certificate scope, validity dates, and device scope must align precisely across both parties. Discrepancies block registration and delay market access.
Market Surveillance module
The Market Surveillance module captures post-market data submitted by competent authorities and, in some cases, manufacturers. It records field safety corrective actions, non-conformities, and surveillance outcomes. This module is mandatory from May 28 and requires cross-functional ownership of product data across regulatory, quality, and production teams to maintain accurate, continuous reporting.
Vigilance and Clinical Investigations modules
These two modules are not mandatory on May 28. Mandatory use for Vigilance and Clinical Investigations is anticipated around 2027. That said, manufacturers running clinical investigations or managing serious incident reports should treat these modules as active preparation priorities now. The data structures, translation requirements, and reporting workflows are complex and not deployable overnight.
Translation requirements for EUDAMED submissions
EUDAMED does not operate in one language. Device labeling data, vigilance reports, and clinical investigation summaries must be accurate across the official EU languages relevant to each market. This is where many manufacturers encounter a compliance gap they did not anticipate.
The practical translation obligations break down as follows:
Device labeling data: UDI-related fields, intended purpose statements, and instructions for use must reflect the same terminology in every submitted language. A mistranslation in the intended purpose field is not a formatting error. It is a regulatory misrepresentation that can trigger rejection or a conformity challenge.
Vigilance reports: Serious incident reports submitted through EUDAMED require precise language. A mistranslated severity descriptor or causal relationship statement can result in a rejected report or a delayed regulatory response, both of which carry audit consequences.
Clinical investigation summaries: For manufacturers already working in the Clinical Investigations module, multilingual summaries must maintain scientific and regulatory accuracy across all submitted languages. Errors in these documents affect both the study record and downstream regulatory decisions.
EUDAMED-grade translation requires adherence to ISO 17100 for translation quality, ISO 13485 for medical device quality management alignment, and ISO 27001 for information security. These are not optional certifications for regulated submissions. They are the controls that make a translation auditable and defensible.
Standard machine translation (MT) produces literal output with weak context handling, which creates a high risk of meaning errors in safety-critical text. Neural machine translation (NMT) engines available as consumer SaaS tools offer better fluency but inconsistent terminology control and limited governance for regulated documentation. Neither approach satisfies audit requirements without additional controls.

AD VERBUM’s AI+HUMAN hybrid translation addresses this through a four-stage workflow. First, client Translation Memories and Term Bases are ingested to constrain output to approved terminology. Second, the proprietary LLM-based LangOps System generates target language output within those constraints. Third, a certified subject-matter expert reviews for technical accuracy, regulatory compliance, and contextual nuance. Fourth, QA is applied under ISO 17100, ISO 18587, and MDR-relevant sector requirements.
Pro Tip: Before uploading any device labeling data to EUDAMED, run a terminology audit against your existing Term Base. Inconsistent terminology between your technical file and your EUDAMED submission is one of the most common causes of registration delays.
For manufacturers managing multilingual clinical trial risks across EU sites, the translation governance requirements are equally demanding. Unvetted translations in regulated submissions create liability that extends beyond the submission itself.
Step-by-step preparation for the May 28 deadline
The EUDAMED mandatory requirements do not reward last-minute preparation. The following sequence reflects the practical order of operations for manufacturers approaching the May 28 deadline.
Audit and remediate internal product master data. Map every device in your portfolio against EUDAMED’s required data fields: Basic UDI-DI, EMDN code, intended purpose, classification, and technical dimensions. Identify gaps in your ERP or quality management system and assign cross-functional ownership to resolve them. This step alone can take months.
Register actors and obtain SRNs immediately. Actor registration is the prerequisite for every other module. Submit your organization registration, designate at least two LAAs, and confirm SRN issuance. Do not proceed to device registration without confirmed SRN status.
Sequence device data uploads by risk class. Prioritize Class III and Class IIb devices first, as these carry the highest regulatory scrutiny. Work through Class IIa and Class I devices in parallel where capacity allows. Confirm alignment with your Notified Body on certificate data before upload.
Establish certified translation workflows for all EUDAMED-required documents. Identify which documents require multilingual submission: labeling data, intended purpose statements, vigilance report templates, and clinical summary formats. Contract certified translation under ISO 17100 and ISO 13485 alignment before the deadline, not after.
Coordinate directly with your Notified Body. Confirm that your certificate data in EUDAMED matches what your NB will upload. Request confirmation of their EUDAMED submission schedule and resolve any scope discrepancies in advance.
Test your vigilance reporting workflow now. Even though the Vigilance module is not mandatory until approximately 2027, your internal process for generating, translating, and submitting serious incident reports should be validated before it is legally required.
Pro Tip: Assign a dedicated EUDAMED data owner in your regulatory team with authority to approve submissions across departments. Organizations that treat EUDAMED as a shared responsibility without a single owner consistently experience data inconsistencies that block registration at the worst possible time.
EUDAMED compliance readiness requires treating this as a cross-departmental program, not a regulatory team task. IT, quality, production, and regulatory must operate from the same data governance framework.
Failure modes and how to mitigate them
The impact of EUDAMED on compliance is most visible when things go wrong. These are the failure patterns that appear most frequently and the controls that prevent them.
Incomplete actor registration blocking all downstream activity. If your SRN is not confirmed before May 28, no device registration is possible. Importers and distributors are obligated to verify EUDAMED registration before placing devices. A registration gap means immediate stock obsolescence and sales prohibition.
Single LAA coverage creating access loss. If your only LAA leaves the organization or becomes unavailable, your entire EUDAMED account is frozen. The last active LAA cannot be terminated, which means recovery requires direct intervention from the competent authority. Maintain two active LAAs at all times.
Data inconsistencies blocking UDI registration. Fields such as EMDN codes, device dimensions, and intended purpose statements must match exactly across your technical file, your quality management system, and your EUDAMED submission. A single field mismatch can block the entire device record.
Uncertified translations triggering rejected vigilance reports. A vigilance report submitted with a translation that misrepresents severity or causality is not just a quality failure. It is a regulatory event that can result in fines and enforcement action.
Delayed implementation creating revenue exposure. Non-compliance triggers immediate market exclusion across all product lines, not just the affected device. This is not a proportionate penalty. It is a system-level hard stop.
“EUDAMED’s supply chain hard stop makes registration compliance a de facto market entry requirement in Europe. Importers and distributors cannot legally place non-registered devices, which means a manufacturer’s registration failure becomes a supply chain failure within days.”
For manufacturers concerned about EU technical data risks from unvetted translation providers, the audit exposure from a rejected submission is compounded by the difficulty of demonstrating due diligence after the fact.
My perspective on what this deadline actually demands
I’ve worked with regulatory teams across medical device manufacturers for years, and the pattern I see with EUDAMED is consistent. Organizations treat it as a documentation project until they realize it is a data infrastructure project. By then, the timeline is compressed.
The shift from voluntary to mandatory use is not a compliance checkbox. It is a market access milestone. If your devices are not registered in EUDAMED by May 28, they cannot legally be sold in the EU. That is a revenue event, not a regulatory finding.
What I’ve found is that the organizations that manage this well share one characteristic: they started data remediation early and assigned cross-functional ownership before the deadline pressure arrived. The ones that struggle are still debating which team owns the EMDN code field in April.
On translation specifically, I’ve seen manufacturers assume that their existing translation vendor can handle EUDAMED submissions because they’ve handled IFU translations before. The certification stack is different. ISO 17100, ISO 13485, and ISO 27001 together are not a standard translation agency offering. They are a regulated-industry requirement, and the difference shows up in audit.
My advice: treat May 28 as a business continuity date, not a compliance date. The distinction changes how you resource it.
— Viestarts
How AD VERBUM supports EUDAMED-grade compliance
EUDAMED submissions require translation that is certified, auditable, and consistent across every language your devices are registered in. AD VERBUM holds ISO 17100, ISO 13485, and ISO 27001 certifications, which together constitute the certification stack that EUDAMED-grade translation requires. The AI+HUMAN hybrid workflow integrates your existing Translation Memories and Term Bases, applies the proprietary LangOps System for terminology-governed output, and delivers certified subject-matter expert review under MDR-aligned QA. Turnaround runs three to five times faster than traditional workflows, and all processing runs on private EU-hosted infrastructure with no public cloud exposure. For manufacturers approaching the May 28 deadline with labeling data, vigilance report templates, or clinical summaries requiring certified medical device localization, AD VERBUM provides the compliance controls your submissions depend on. Contact us for a compliance translation assessment tailored to your EUDAMED submission scope.
FAQ
What modules become mandatory on May 28, 2026?
The Actors, UDI/Devices, Notified Bodies and Certificates, and Market Surveillance modules become mandatory on May 28, 2026. The Vigilance and Clinical Investigations modules are expected to follow around 2027.
What happens if a manufacturer is not registered in EUDAMED by May 28?
Non-compliance triggers immediate market exclusion. Importers and distributors cannot legally place unregistered devices, which means sales prohibition takes effect across affected product lines without a grace period.
How many Local Actor Administrators does an organization need in EUDAMED?
Each organization must maintain at least two Local Actor Administrators. The last active LAA cannot be terminated, and losing sole LAA access requires competent authority intervention to recover.
What translation certifications are required for EUDAMED submissions?
EUDAMED-grade translation requires ISO 17100 for translation quality, ISO 13485 for medical device quality management alignment, and ISO 27001 for information security. These certifications make submissions auditable and defensible under MDR.
How long does data remediation take before EUDAMED submission is possible?
Data cleaning and alignment of internal ERP and product master data can take 12 to 18 months. Many systems lack the structured fields EUDAMED requires, such as EMDN codes, making early remediation the single most important preparation step.
Sources:
EUDAMED four first modules will be mandatory use 28 May 2026 — European Commission
The Business Impact of EUDAMED on Market Access — MedEnvoy
Business Impact of EUDAMED and Staying Compliant — MedEnvoy
EUDAMED compliance readiness — CE Interim
Managing actors and user accounts — EUDAMED Information Centre
Localization — AD VERBUM
Recommended