
Master post-market surveillance translation under EU MDR
- Mar 20
- 10 min read

Many regulatory professionals assume that translating post-market surveillance documentation is straightforward, a box-ticking exercise that can be handled by general translation services. This misconception leads to compliance gaps that surface during notified body audits, competent authority inspections, and field safety corrective actions. Under EU MDR 2017/745, post-market surveillance obligations generate a sustained multilingual documentation burden across PMCF plans, periodic safety update reports, and field communications. Terminology inconsistency creates traceability risk across the product lifecycle. This guide outlines the PMS document categories generating highest translation risk, the notified body review patterns that identify translation-induced ambiguity, and how ISO 13485-aligned workflows with medical device SME review address the surveillance documentation standard.
Table of Contents
Understanding Post-Market Surveillance Documentation Under EU MDR
Common Translation Pitfalls In Post-Market Surveillance Documentation
Best Practices For Compliant Multilingual Post-Market Surveillance Documentation
Regulatory Oversight And Multilingual Documentation Compliance: What Notified Bodies Scrutinize
Key takeaways
Point | Details |
PMS translation is mandatory | EU MDR requires accurate multilingual versions for all markets where devices are sold. |
Terminology consistency is critical | Inconsistent terms across PMS documents create traceability failures during audits. |
Notified bodies scrutinize translations | Auditors verify accuracy, completeness, and proper version control in all language versions. |
ISO 13485 workflows reduce risk | Structured translation processes with SME review ensure regulatory compliance. |
Update cycles demand retranslation | Every PMS revision must trigger synchronized multilingual updates across all markets. |
Understanding post-market surveillance documentation under EU MDR
Post-market surveillance documentation is a mandatory component under EU MDR requiring detailed reporting and updates. PMS encompasses systematic activities manufacturers conduct to gather, record, and analyze data about the safety and performance of devices already on the market. This includes post-market clinical follow-up plans, periodic safety update reports, trend reports, and field safety corrective action communications.
EU MDR Article 83 establishes the PMS system requirement. Manufacturers must document their surveillance activities, maintain records of complaints and adverse events, and produce regular reports for notified bodies and competent authorities. These documents form the evidence base demonstrating ongoing compliance with essential safety and performance requirements.
Why must PMS documents be multilingual? Regulatory authorities in each EU member state require submissions in their official language. Notified bodies conducting conformity assessments need translations to verify compliance. Distributors, importers, and healthcare professionals require MDR technical documentation in local languages to fulfill their own regulatory obligations. Field safety notices must reach end users in languages they understand to ensure patient safety.
Inaccuracies or omissions in translations create direct compliance risks:
Misinterpreted safety signals leading to delayed corrective actions
Terminology mismatches between PMS reports and technical files causing traceability failures
Incomplete translations of adverse event descriptions compromising vigilance reporting
Legal liability exposure when field safety notices contain translation errors
Audit findings and potential certificate suspension when notified bodies identify translation deficiencies
The EUDAMED database adds another layer of complexity. Manufacturers must upload PMS data and reports to the European database, with specific fields requiring translation into multiple languages. Version control becomes critical when updating both the database and distributed PMS documents simultaneously across language versions.
Pro Tip: Establish a master glossary of device-specific and regulatory terms before beginning any PMS translation project. This glossary should align with terminology used in your technical documentation, instructions for use, and previous regulatory submissions to ensure consistency across your entire documentation ecosystem.
Common translation pitfalls in post-market surveillance documentation
Many manufacturers underestimate the complexity of translation requirements, leading to compliance failures. The most frequent mistake is treating PMS translation as a simple linguistic conversion rather than a regulatory compliance activity requiring medical device expertise.
Incomplete translations represent a widespread problem. Manufacturers often translate the main body of PMS reports while leaving appendices, data tables, or technical annexes in the source language. Notified bodies and competent authorities reject submissions with partial translations, causing delays and additional costs. Some manufacturers mistakenly believe that English versions suffice for all EU markets, ignoring national language requirements in countries like France, Germany, and Spain.
Failing to update translated documents alongside PMS report revisions creates version control chaos. When manufacturers issue revised periodic safety update reports or updated PMCF plans, they must simultaneously update all language versions. Delays in translation create situations where different markets operate with different information, a serious compliance violation that undermines the entire PMS system.

Cultural and linguistic nuances significantly affect regulatory acceptance. Direct translations of adverse event descriptions may not convey the same clinical meaning across languages. Legal terminology varies between jurisdictions, and what constitutes adequate disclosure in one language may fall short in another. Regulatory expectations for documentation style, formatting, and level of detail differ between member states.
Overlooking the need for certified translations in specific EU countries creates additional problems. Some competent authorities require sworn translations or translations bearing specific certifications. Without understanding these jurisdiction-specific requirements, manufacturers face submission rejections.
Translation errors impact audit outcomes directly. During conformity assessments, notified bodies compare language versions of PMS documents against technical files and other regulatory submissions. Terminology inconsistencies raise red flags. Mistranslated safety information suggests inadequate quality management. Incomplete translations indicate systemic compliance weaknesses. These findings can delay certificate renewals or trigger additional scrutiny of the entire quality system.
Market access suffers when translation deficiencies surface. Competent authorities may suspend device registrations pending correction of documentation issues. Distributors refuse to handle products without proper local language documentation. Healthcare facilities reject devices lacking appropriate PMS documentation in their national language.
Pro Tip: Create a translation impact assessment matrix that maps each PMS document type to its regulatory touchpoints. Identify which documents go to notified bodies, competent authorities, distributors, and end users, then prioritize translation resources based on regulatory risk and compliance deadlines.
Best practices for compliant multilingual post-market surveillance documentation
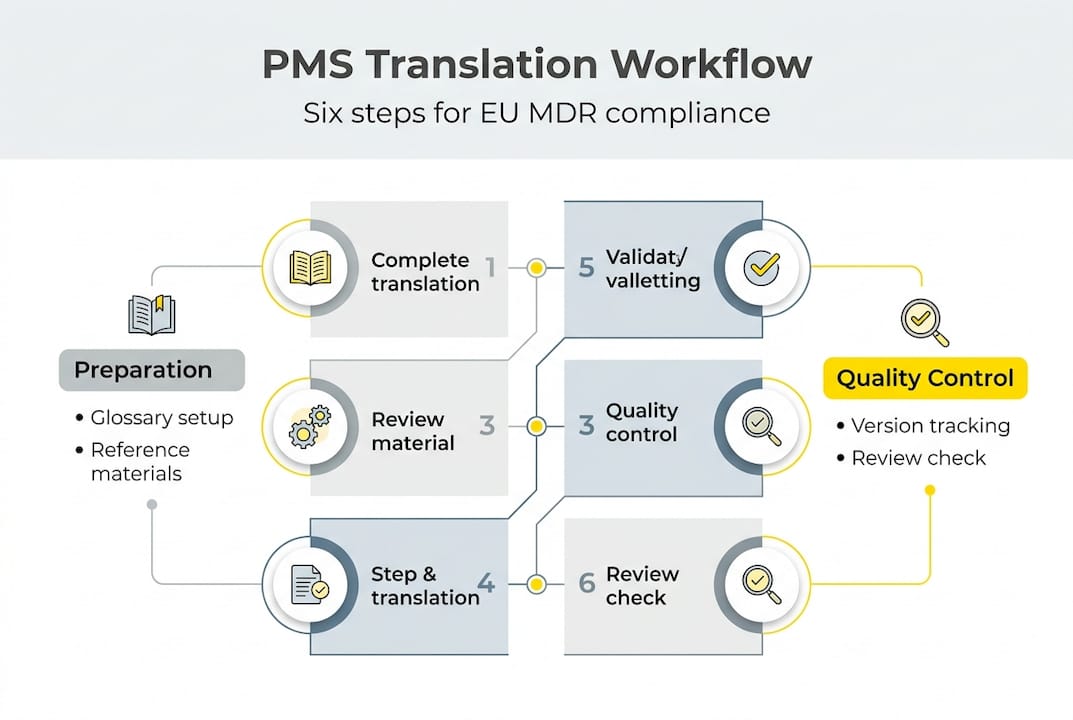
Following standardized workflows for regulated document translation reduces risk and improves compliance. Implement a structured translation process specifically designed for PMS documentation under EU MDR.
Step 1: Establish a PMS translation governance framework. Designate a responsible person within your quality management system to oversee all PMS translation activities. Define clear procedures for initiating translation requests, selecting language service providers, reviewing translations, and approving final versions. Document these procedures in your quality manual.
Step 2: Select qualified language service providers with demonstrated MDR expertise. Verify that providers employ medical device subject matter experts, maintain ISO 13485 certification, and follow ISO 17100 translation standards. Request evidence of previous PMS translation projects and ask for references from other medical device manufacturers.
Step 3: Provide comprehensive reference materials to translators. Supply your master glossary, previous regulatory submissions, approved translations of technical documentation, and relevant standards. The more context translators have, the more accurate and consistent their output.
Step 4: Implement routine updates and version control for multilingual content. When you revise any PMS document, immediately flag all language versions for update. Use a document management system that tracks version numbers, revision dates, and approval status across all languages. Never allow different language versions to fall out of sync.
Step 5: Ensure legal and cultural validation of translations. Have in-country regulatory experts review translations for compliance with local requirements. Verify that legal terminology aligns with national regulations and that clinical descriptions match local medical practice standards.
Step 6: Conduct quality reviews before submission. Compare translated PMS documents against source documents using qualified reviewers who are fluent in both languages and understand medical device regulations. Check for completeness, accuracy, consistency with other documentation, and adherence to formatting requirements.
Tools and technologies assist in compliant translation management. Translation memory systems store previously translated segments, ensuring consistency across documents and reducing costs for updates. Terminology management databases enforce use of approved terms. Workflow management platforms track translation projects from initiation through approval, maintaining audit trails required by ISO 13485.
Translation Stage | Key Activity | Quality Check |
Preparation | Glossary creation, reference material assembly | Completeness verification |
Translation | Subject matter expert translation | Terminology consistency |
Review | Bilingual expert comparison | Accuracy confirmation |
Validation | In-country regulatory review | Local compliance check |
Approval | Quality management sign-off | Final release authorization |
Pro Tip: Build translation timelines into your PMS planning cycles. If you know you must submit periodic safety update reports annually, schedule translation activities 60 days before submission deadlines to allow adequate time for translation, review, and any necessary revisions.
Regulatory oversight and multilingual documentation compliance: what notified bodies scrutinize
Notified bodies emphasize accuracy, completeness, and traceability of translated PMS documentation in audits. Understanding their evaluation criteria helps manufacturers prepare compliant multilingual documentation.
Notified bodies assess whether translated PMS documents convey the same information as source documents without loss of meaning or introduction of ambiguity. They verify terminology consistency between PMS reports and other elements of the technical documentation. They check that safety-critical information receives particular attention in translation, with no softening or strengthening of risk statements.
Auditors examine documentation traceability across language versions. They compare device identifiers, reference numbers, dates, and version codes to ensure alignment. They verify that translated documents cite the same standards, regulations, and clinical literature as source documents. They check that data tables, charts, and figures maintain identical values across all languages.
Transparency expectations are high. Notified bodies want to see documented translation processes within your quality management system. They review procedures for translator qualification, translation review, and approval of multilingual documents. They examine records showing who translated each document, who reviewed it, and who approved it for regulatory use.
Key criteria notified bodies use:
Completeness: All sections, appendices, and annexes translated without omissions
Accuracy: Technical and clinical terminology correctly rendered in target languages
Consistency: Alignment with terminology used in technical files and previous submissions
Traceability: Clear version control and document identification across languages
Timeliness: Translated versions available when required for submissions and audits
Compliance Failure | Best Practice |
Partial translations with untranslated sections | Complete translation of all document elements |
Inconsistent terminology across PMS documents | Master glossary enforced across all translations |
Missing version control in translations | Document management system tracking all language versions |
Unqualified translators without medical device expertise | Subject matter expert translators with regulatory knowledge |
No documented translation review process | Formal review and approval procedures in quality system |
Notified bodies pay special attention to field safety corrective actions and related communications. These documents directly affect patient safety, so translation accuracy is paramount. Auditors verify that urgency and severity of safety issues come through clearly in all language versions. They check that corrective action instructions are unambiguous and actionable in each target language.
During EUDAMED 2026 compliance reviews, notified bodies examine how manufacturers manage multilingual data entry and reporting. They verify that information uploaded to the database matches information in distributed PMS documents across all languages.
Manufacturers who demonstrate robust translation quality management, documented procedures, qualified resources, and systematic verification processes typically navigate notified body audits successfully. Those who treat translation as an afterthought face findings, corrective action requests, and potential compliance issues that delay or jeopardize market access.
How AD VERBUM supports your EU MDR translation needs
Navigating the complex multilingual requirements of post-market surveillance documentation demands specialized expertise and proven processes. AD VERBUM brings 25 years of experience in regulated sector translation, with deep knowledge of EU MDR requirements and ISO 13485 quality standards. Our proprietary AI+HUMAN hybrid translation workflow combines advanced language technology with certified medical device subject matter experts to deliver accurate, compliant PMS documentation.
Our network of 3,500+ specialized linguists includes medical professionals, regulatory experts, and engineers who understand both the technical content and regulatory context of PMS documents. We maintain ISO 13485 certification and align our quality processes with ISO 17100 and ISO 18587 standards. Our EU-hosted infrastructure ensures data sovereignty and GDPR compliance for sensitive device documentation. Whether you need professional translation services for periodic safety update reports, multilingual SEO services to optimize your regulatory content, or comprehensive localization services for field safety communications, AD VERBUM delivers the precision and compliance assurance your post-market surveillance program requires.
FAQ
What are the key translation requirements for post-market surveillance under EU MDR?
PMS documents must be translated accurately into all languages of the EU countries where the device is marketed, ensuring legal terminology is precise and validated. This includes PMCF plans, periodic safety update reports, trend reports, and field safety corrective action communications. Translations must maintain terminology consistency with technical documentation and previous regulatory submissions. Competent authorities and notified bodies require complete translations, not partial or summary versions, to fulfill their oversight responsibilities.
How often should post-market surveillance documentation be updated and retranslated?
PMS documentation must be updated and translated promptly whenever significant changes occur, typically aligned with periodic review cycles mandated by EU MDR. For most devices, this means annual updates at minimum, with immediate updates required when new safety information emerges or corrective actions are implemented. Every source document revision triggers a corresponding translation update across all language versions. Maintaining synchronized versions across languages is a core compliance requirement that notified bodies verify during audits.
What role do notified bodies play in reviewing multilingual PMS documentation?
Notified bodies verify translation accuracy, completeness, and proper documentation control to ensure that PMS reports meet regulatory standards across all EU languages. During conformity assessments and surveillance audits, they compare language versions against each other and against other technical documentation to identify inconsistencies or gaps. They assess whether your quality management system includes adequate procedures for managing multilingual documentation. Translation deficiencies can result in audit findings, corrective action requests, or delays in certificate renewals.
Why is terminology consistency critical in PMS translations?
Inconsistent terminology across PMS documents and other regulatory submissions creates traceability failures that undermine your entire compliance posture. When the same device component or adverse event is described differently in various documents or language versions, auditors cannot verify that you are discussing the same thing. This breaks the chain of evidence linking clinical data, risk assessments, and post-market experience. Notified bodies view terminology inconsistency as a quality system weakness that may indicate inadequate document control or insufficient attention to regulatory requirements.
How do I select a qualified translation provider for MDR compliance?
Look for providers with demonstrated medical device expertise, ISO 13485 certification, and adherence to ISO 17100 translation standards. Verify that they employ subject matter expert translators with regulatory knowledge, not just linguistic skills. Ask for references from other medical device manufacturers and examples of previous PMS translation projects. Evaluate their quality management processes, including how they handle terminology management, version control, and translation review. Ensure they understand jurisdiction-specific requirements and can provide in-country regulatory validation when needed.
Recommended

